
Shaojun TANG
BSBE
SARS-CoV-2 Spike Protein Post Translational Modification Landscape and Its Impact on Protein Structure and Function via Computational Prediction

To elucidate the role of post-translational modifications (PTM) in SARS-CoV-2 spike protein’s structure and virulence, we generated a high-resolution map of 87 PTMs using liquid chromatography with tandem mass spectrometry (LC-MS/MS) data on extracted spike protein from the SARS-CoV-2 virions, and then we proposed the mutagenesis approach to mimic the influence of PTMs on protein structure due to altered physicochemical properties in modified amino acids, and then reconstituted the spike protein’s structure from the substituted sequences. For the first time, the proposed method revealed predicted protein structures resulting from PTMs, a problem that Alphafold2 has yet to address. In addition, we also performed computational analyses of the interaction of post-translationally modified spike protein with its host factors such as ACE2 to illuminate the binding affinity. The workflow of the study is shown in Figure 1.
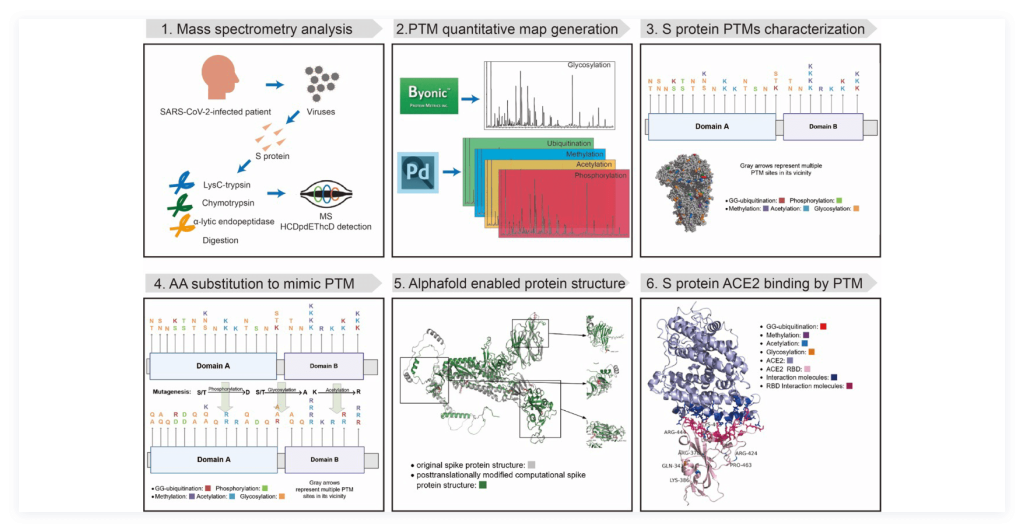
1) Construction of the high-resolution quantitative map of spike protein PTMs: We generated a high-resolution map of 87 PTMs using liquid chromatography with tandem mass spectrometry (LC-MS/MS) data on extracted spike protein from the SARS-CoV-2 virions. Particularly, we identified 14 PTMs in the RBD region of the spike protein S1 unit, including 3 glycosylation sites, 4 methylation sites, 2 ubiquitination sites, and 5 acetylation sites, as is shown in Figure 2.

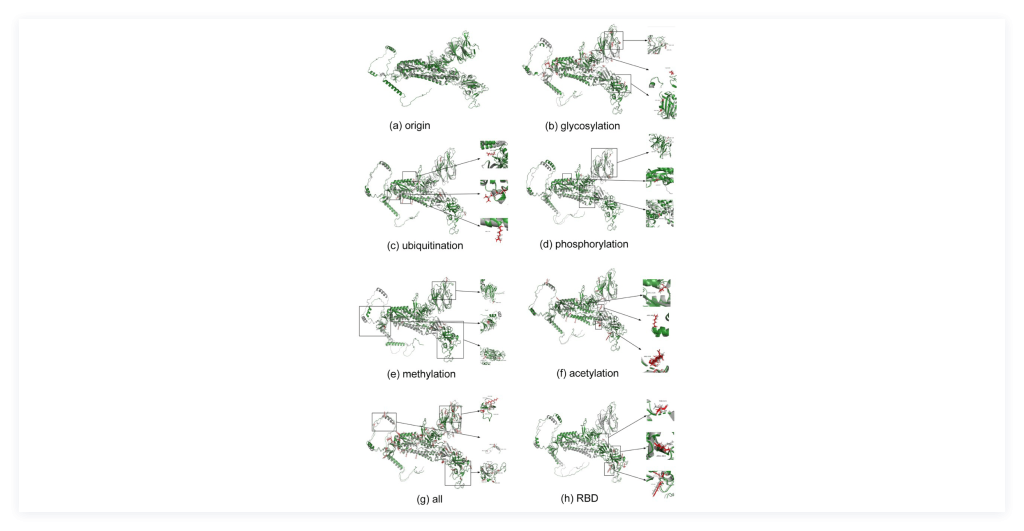
2) In silico site-directed mutagenesis to derive substituted amino acid sequences on PTM sites: Mutagenesis is a popular strategy to study protein modifications by replacing an amino acid with another amino acid to understand how the consequence of a PTM in terms of residue change impacts the protein structure and function. We proposed the mutagenesis approach: in-silico, site-directed amino-acid substitution to mimic the influence of PTMs on protein structure due to altered physicochemical properties in modified amino acids, and then reconstituted the spike protein’s structure from the substituted sequences. Finally, we performed comparison of protein structures from computational prediction and Cryo-EM experiment to evaluate and validate the accuracy of predicted spike protein structure, as is shown in Figure 3.
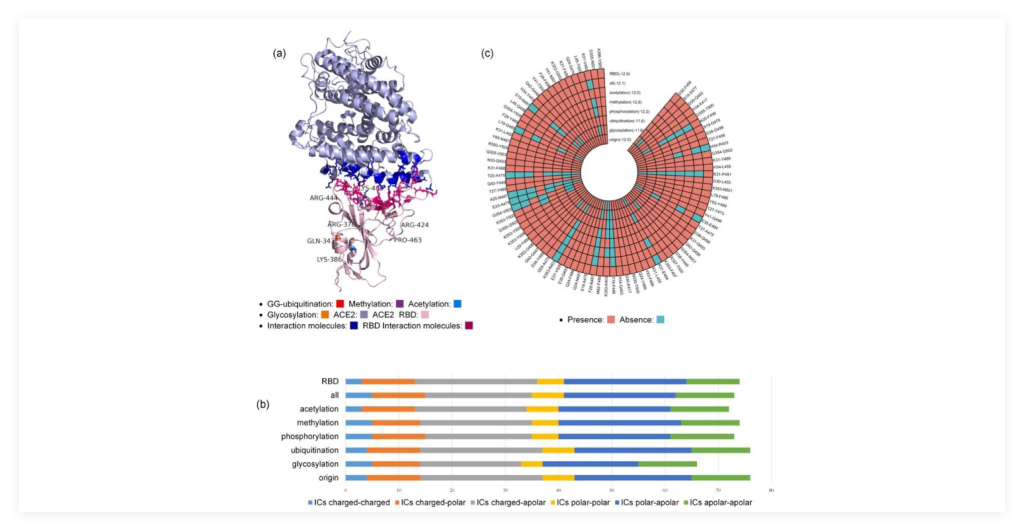
3) Computational prediction of PTM on the binding affinity between SARS-CoV-2 spike protein RBD region and human host factors: We used computational approaches to analyze the virion spike protein and host factor binding interaction by evaluating the impact of post-translational modifications as shown in Figure 4. For instance, different PTM types have varying impact on the change of protein structure, with methylation having the largest effect and ubiquitination having the smallest effect.
Conclusion: Overall, we characterized a total of 87 PTM sites on 5 major medication types in the SARS-CoV-2 spike protein, many of the PTMs are novel. Subsequently, we proposed and validated a computational approach to predict the spike protein’s structures from widely studied PTMs using in-silico mutagenesis and Alphafold2. Results showed substantial changes in spike protein’s structures, especially in RBD domain, as a result of amino acid modifications. Our study suggests that virulence is partially explained by the PTMs. In summary, in the absence of costly and laborious method for characterizing protein structural changes due to PTMs, we believe that our proposed innovative computational algorithm can pinpoint an effective approach for protein structural characterization and functional study. Nevertheless, these results require further validation by experimental X-ray crystallography and/or Cryo-EM.